Open Theses Topics
Current suggestions for B.Sc. / M.Sc. / Ph.D. theses topics. Should you have an idea for a topic yourself, don't hesitate to contact us to discuss it.
The bioinformatics masters curriculum contains two lab rotations (Laborpraktika). Students shall independently work on a e.g. programming or analysis project to experience necessary skills prior to their masters thesis. We always welcome interested students and have plenty of topics; some examples are listed here. Please get in contact with us!
For Hackers: make spheres smooth again |
|
|---|---|
 |
Many microbiome publications contain Emperor plots. Mine always do :-) Unfortunately, exporting graphics from Emperor come with some SVG artifacts, which makes icons quite ugly (see the accoring github issue). This seems to be base in some floating point disagreements based in the underlying THREE.js library. The non trivial, very technical task for this thesis is to update the THREE.js library of Emperor to a more recent version. |
Caterpillars as a replacement for mammalian models in preclinical research |
|
 © Fraunhofer IME | Kim Weigand |
Insect larvae such as tobacco hornworm can accelerate and economize preclinical research by complementing classic laboratory animals such as rats and mice. Small mammals like mice or rats are indispensable for preclinical research. However, growing ethical concerns led to the incorporation of the 3R principle (replacement, reduction, and refinement) into animal experiments legislation and research funding. In the future, the number of vertebrate laboratory animals should be reduced, and non-vertebrate alternatives should be used where possible. Furthermore, the incorporation of the 3R principle will also economize preclinical research since insect husbandry is much cheaper than the traditional housing of laboratory mammals. In that context, insect larvae like Manduca can serve as an alternative in vivo animal model. Mainly, with the high degree of evolutionary conservation in the innate immunity of the gut and similarities in the enteric epithelial structure, Manduca sexta can serve as a model for human gut inflammation. Recently we employed larvae of the tobacco hornworm Manduca sexta, which are big enough for macroscopic imaging techniques, such as computed tomography (CT), magnetic resonance imaging (MRI), and positron emission tomography (PET) as a high-throughput platform to study the innate immunity of the gut and host-pathogen interactions. The developed platform represents an ethically acceptable, resource-saving, large-scale, and 3R-compatible screening tool for various life science disciplines, including the identification of new effectors and inhibitors in gut inflammation, the assessment of pesticides or other environmental factors, the assessment and evaluation of new antibiotic therapies, the analysis of host-pathogen interactions, and the identification of new contrast agents or tracers in radiology. Since 75% of the known human disease-causing genes have homologs in insects, this approach will also be helpful in testing preclinical hypotheses in inflammatory bowel disease. The Problem: Further reading:
|
Tiny tourists or blind passengers? - Determining the microbial diversity of the Cologne-Bonn-Airport. |
|
 |
Join us on a fascinating journey into the microbial world lurking within one of Germany's busiest travel hubs, the Cologne-Bonn Airport. We invite you to delve into the captivating realm of microorganisms and their potential impact on passenger health and aviation safety. In recent years, technological advancements have revolutionized the way we study microorganisms. High-throughput methods now enable us to explore the vast diversity of microbial genomes that inhabit both hosts and environments. As air travel continues to soar, with over three billion passengers taking to the skies annually, understanding the risks associated with in-flight transmission of infectious diseases has become a paramount global health concern. Numerous documented cases, such as influenza, meningococcal infections, norovirus, SARS, and multi-drug resistant tuberculosis, highlight the potential for disease transmission during air travel. Research on SARS and pandemic influenza has revealed that airplanes can serve as rapid conduits for the spread of emerging infections and pandemics. Moreover, studies suggest that passenger and crew movements, as well as their close contacts, play a crucial role in disease transmission dynamics. This master thesis project will focus on the critical field of microbial monitoring at the Cologne-Bonn Airport, specifically examining highly frequented locations such as stairways and escalators. By thoroughly investigating the microbial diversity within these areas, we aim to shed light on the potential health risks associated with air travel and contribute to the development of effective aviation safety measures. By embarking on this groundbreaking research endeavor and the corresponding comprehensive analysis, we aim to provide valuable insights and recommendations that can enhance the safety and well-being of travelers. Help us to unlock the secrets of the airport microbiome and ensure aviation safety! ? sign now |
Refactor my quick but dirty ggmap microbiome analysis collection. |
|
 https://bonkersworld.net/building-software |
Back in 2016, I realized that I copy & pasted code snippets for my various microbiome analysis. Instead of coming up with a clean concept to export these snippets, I abused another repository (hence the strange name for mapping identifiers from GreenGenes) to at least re-use the same code: ggmap. It once even had some unit tests, but in order to go fast, I ignored them failing over time. I never intended someone else to use this uggly part of software, but now realize that I torture my students by forcing them to do exactly this: use this badly documented, wobbly, partially deprecated code collection. Should you have a slightly masochistic fun in refactoring my python code, I (and probably my students) would greatly appreachiate your efforts. |
Hunt for exoribonuclease-resistant RNA |
|
 |
The exoribonuclease-resistant RNA has some fascinating biological functions established through a very specific confirmation including a pseudoknot. This feature makes it a hard task to search for homologs. However, we should be able to design a hand tailored RNA folding algorithm for this pseudoknotted motif and search for compatible sequences in multiple genomes. (keywords: TDM for pknotsRG, alignment in Rfam: https://rfam.org/family/PLRV_xrRNA#tabview=tab9) |
RNAshapes studio front-end |
|
 |
Many RNA molecules do not encode proteins (mRNA), but by themselves exert important biological functions. Most functions are realized through their three dimensional structure - which is often times more conserved than the primary nucleotide sequence. Thus, sequence homology search with e.g. BLAST often does not return good results. Secondary structure of RNA is the set of nucleotides that form base-pairs and functions as a scaffold for the final 3D structure. Since RNA folds hierarchically, prediction of secondary structure gives valuable insights about an RNA - while true 3D predictions are mostly computational intractable. RNAshapes / pKiss / KnotInFrame and other software packages were developed at Bielefeld University and are popular tools to predict different aspects of secondary structures. They are based on the same algorithmic ideas (algebraic dynamic programming) and share the same code base back-end. The front end is written in Perl and lacks modern software engineering must-haves, like continuous testing, modularity or documentation. Since our group is working on several algorithmic extensions of the back-end, we are looking for an encouraged student who re-implements the front-end in Python3, adds unit tests, creates bioconda packages, ... to allow for easy maintenance and distribution. |
Quantify folding ensemble differences |
|
 https://pixabay.com/photos/tape-measure-measure-up-2734127/ |
Stub: Bray-Curtis of dot-plot for related RNA sequences + UniFrac with tree that reflects neighborhood? |
UTF8 for RNA |
|
 |
Bellman's GAP cannot parse non-ASCII bases, i.e. base modifications. But those become ever more important, like pseudouridine in BioTechs Covid19 vaccine. There are a few thermodynamic parameters out there, that might be worth being integrated into Vienna's parameters + enable UTF8 parsing for Bellman's GAP. |
What's in my sample |
|
 https://www.nnkgreen.org/tracking-the-wild-woozle-event |
stub: machine learning on EMP dataset to guess metadata values for novel microbial samples, or "enrich" metadata. |
Non-Terminal Report Algebra |
|
| Similar to the automatic generation of "enum" algebras, one could think of automatic algebras that report non terminal use. Could be handy for debugging of outside code generation. | |
Bioconda for FlowSoFine |
|
 |
Create two (bio)conda packages for the R software FlowSoFine and its app. |
TAKEN: Nextflow Pipeline improvements |
|
 |
Are you interested in learning reproducibility and a modern workflow language hands on? Are you interested in visualization and like paying attention to detail? Then help us improve our human genomics filtering pipeline! The pipeline is written in Nextflow and uses Podman containers to ensure reproducibility. It filters out human genomic information from metatranscriptome samples taken from humans. For this purpose it maps against multiple references. It is important to filter human reads to ensure that the human host cannot be identified from the remaining genomic data. Your job would be to expand the pipeline by adding visualization steps. We want to show how many reads are filtered out in each step and then show the plots in a nice summary html report. |
TAKEN: The Curious Case of the Caterpillar’s Missing Microbes |
|
 © Fraunhofer IME | Kim Weigand |
Do the caterpillars of Manduca sexta lack a resident gut microbiome?Lately, the idea was challenged that most animals harbor beneficial microbial communities. A prominent study showed that most caterpillars lack a resident gut microbiome. This thesis challenges this idea! We have an extended 16S rRNA Dataset containing gut samples from 16 larvae from different anatomical positions, samples from host plants, and samples from the artificial diet. Your job is to conduct a gut microbiome analysis of the insect larva of Manduca sexta. What we can say for now is that we have found microbes :) Further reading:
|
TAKEN: Transmembrane Prediction via Algebraic Dynamic Programming |
|
 |
Almost 25 years ago, Hidden Markov Models (HMM) were successfully used in the emerging new field of Bioinformatics to predict the topology of trans-membrane proteins (original papers "A hidden Markov model for predicting transmembrane helices in protein sequences" and "Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes") Authors of the program TM-HMM were excited about the tight coupling of mathematical modeling via HMMs and the modeled molecular biology. They used multiple algorithms to "score" amino acid sequences e.g. for being transmembrane proteins (Viterbi) or mark sub-sequences for being the helical regions (forward-backward) transecting the membrane. Labeling the many states of the HMM into "inside", "outside", "transmembrane" or "unlabeled" turns computation of the most probable "label" into an NP-hard problem - as proven much later by Broňa Brejová et al.. The TM-HMM program is a great example for real-world sized instances of dynamic programming; although a modern version is now based on deep learning - which, in principle, also uses forward-backward ideas. Within a thesis, you would need to re-implement the HMM in Algebraic Dynamic Programming such that we can then use novel auto-generated algorithmic ideas to potentially improve prediction accuracy for this long standing problem. |
TAKEN: Speed-Up RNA prediction |
|
 |
We make use of the high abstraction programming discipline "Algebraic Dynamic Programming" (ADP) to provide a wide variety of RNA secondary structure prediction programs (source code, web-services). Our focus was on short development times. Unfortunately, some implementation details - mainly outside the core ADP algorithm - slow down execution of the programs significantly. See image for a profiling example: too much time is spent with unnecessary conversion from and to log-space (red) or repetitive computation of information (magenta) that should better be done prior to the core algorithm. Please give us a hand and benchmark current execution times, make suggestions for speed-ups (there are a lot of low hanging fruits), implement ideas into our ADP compiler and benchmark your improvements as a rewarding thesis! |
TAKEN: AI to predict Leukemia |
|
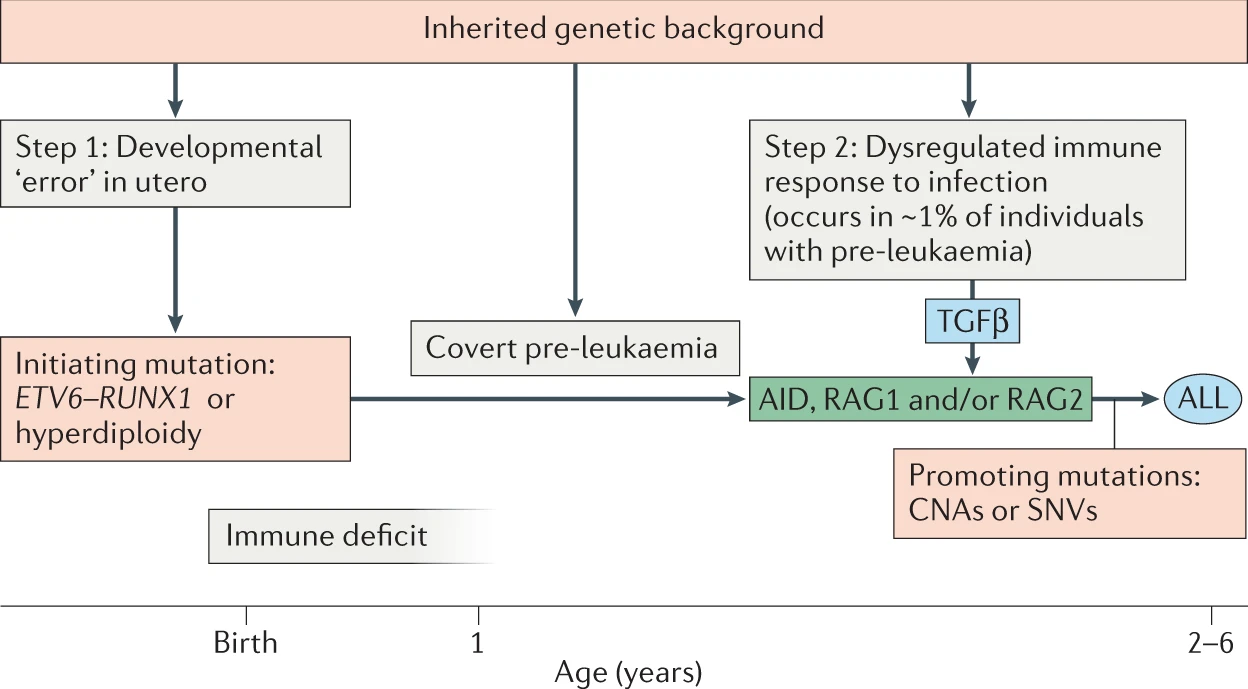 |
The current working hypothesis for the development of Leukemia includes two independent steps. The first is a genetic mutation aquired in utero before birth. A screening showed the alarming fact that 1-5% of all newborn childring carry such a mutations. Fortunately, only a small fraction will develop leukemia - it remains unclear why, i.e. what the triggering factors for the second step is. In a mouse model, we found that the stool microbiome was highly specific for the first step. If we are able to translate this to human we might have a diagnostic tool to identify children at risk. As always, we are lacking data to train such a prediction tool. However, there is a growing number of microbiome experiments that collect these data, e.g. Gut microbiome in pediatric acute leukemia: from prediction to cure. The task of this project would be two-fold: 1) collect microbial data from various experiments 2) use the data to train simple machine learning tools like Random Forest and test if they can predict leukemia suceptibility. |
TAKEN: Ubuntu Package for fold-grammars software |
|
 https://medium.com/@hulkcodexl16/debian-package-development-8edf54ea9e99 |
Create a github action to automatically deploy multiple software packages of the fold-grammars repository (pKiss, RNAshapes, ...) as Ubuntu Launchpad packages, as it was manually done here https://launchpad.net/~bibi-help/+archive/ubuntu/bibitools |
TAKEN: Fungal genome annotation pipeline |
|
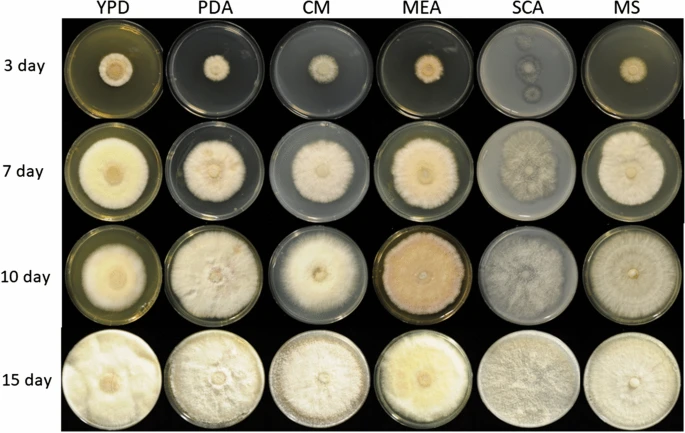 |
The transition to a biobased economy involving the depolymerization and fermentation of renewable agro-industrial sources is a challenge that can only be met by achieving the efficient hydrolysis of biomass to monosaccharides. In nature, lignocellulosic biomass is mainly decomposed by fungi. Further read. In collaboration with the Institute of Food Chemistry and Food Biotechnology, we are sequencing specific fungi genomes and aim to understand their functional capabilities through wet-lab experiments and in-silico genomic annotations. We use Funannotate for the latter, however this pipeline needs to be adapted to operate on our high-performance compute cluster and be specialized towards the fungal genomes for which we generate short- and long- genomic reads and short- transcriptional reads. |
TAKEN: Plotting algebra |
|
| Another use for automatic algebra generation would be for "drawing" candidates ala SVG. | |
TAKEN: QIIME2 wrapper for Dimensionality Reduction techniques |
|
 |
Dimensionality reduction is necessary to project data of microbial experiments into 2D or 3D to allow interactive exploration and hypothesis generation. In QIIME2, one of the leading software platforms for microbial analysis, this is currently supported via Principal Coordinate Analysis (PCoA) with subsequent visualization via the Emperor tool. As with every projection, PCoA cannot preserve all pairwise distances which can generate visual artifacts that mislead the analyst. T-distributed Stochastic Neighbor Embedding (t-SNE) is an alternative to PCoA that focuses on local structures. A similar technique is Uniform Manifold Approximation and Projection (UMAP). |
TAKEN: Bioinformatics of microbial pattern changes detected by flow cytometry |
|
 |
Background At the "Leibniz Research Institute for Environmental Medicine" in Düsseldorf, we used this method and developed a novel and easy approach to evaluate characteristic features and differences between given microbiota samples. In our approach we use hexagonal binning across the bivariate flow cytometry data and the resulting hexagonal gates for dissimilarity calculations. In context of a master thesis our new developed approach should be compared to already established tools for flow cytometry data analysis (FlowEMMi, FlowDiv, ...). Furthermore, the influence of the number of hexagons for automatic and representative clustering as well as for distance matrix of the treatment groups should be analysed. Additionally, correlations analysis with sequencing data are envisaged. Data Requirements Contact |
TAKEN: Mine Qiita |
|
 |
Qiita aggregates over 230,000 publicly available microbiome samples. The majority was created following the Earth Microbiome Project protocol, i.e. using Illumina short read sequencing to obtain V4 amplicon data. These samples from hundreds of studies cover diverse ecosystems and thus capture large portions of bacterial diversity. Many of them are not culturable and don't even have a name. Phylogenetically placing (see "SILVA-SEPP" project) all the sequences (approx. 16,000,000) of those microbiota into the same reference tree, e.g. Greengenes, should reveal interesting trends. All available references are known to be incomplete. Hot spots in the tree, where many different sequences accumulate, would point to potentially novel clades. Grafting de-novo trees of those sequences to the reference might be a practical means to sharpen phylogenetic metrics or suggest sub-sets of original samples that might be worth being subjected to whole genome shotgun sequences to shed light on unseen organisms. The project's aim is to develop a methodology to mine this rich dataset and highlight "interesting" patterns in the phylogenetic reference tree. |
TAKEN: Influence of the genetic background on murine gut microbiome composition and diversity over three generations |
|
 |
The mammal gut is a complex ecosystem harbouring approximately 1014 microorganisms with an important role in health and disease of the host. The main factors that contribute to the inter-individual variation of the intestinal microbiota are the environment, diet, age, gender and genotype. Whether and to which extent the host genotype shapes the gut microbiome is still subject of debate. While some allocate a decisive role to the genotype, others demonstrate that the foster-mother's gut microbiota rather than the genotype are essential in the future composition of the gut microbiota. However, these hypotheses have been drowned secondarily in settings following other objectives and by different methods. Thus, the main goal of this project is to study the drift of gut microbiome of simultaneously, with the same microbiome, naturally colonized C57BL/6J and BALB/c inbreed mice in relation to host genotype, sex and cage. For this C57BL/6J and BALB/c embryos were implanted into B6CF1 recipient foster-mothers in order to obtain parental C57BL/6J and BALB/c mice colonised naturally with the same gut microbiota of the foster-mothers. The two different mice strains were breed completely separated from each other in the standardised environmental conditions of individually ventilated cages (IVCs) over three generations. The standardisation of the environmental conditions will strengthen the role of intrinsic factors such as genotype or sex on microbiome variation. From the parental as well as from each of the three following generations, the composition of gut microbiome was recorded at 7 and 15 weeks of age by next-generation-sequencing analysis of V3-V4 regions of the 16S rRNA genes isolated from fecal and cecal samples. This 16S analysis project requires to apply multiple alpha- and beta-diversity metrics to quantify complexity and diversity of the different microbial communities. Furthermore, differentially abundant features must be identified via discrete false discovery rates or similar statistical tools. These analyses among the two genetic backgrounds will allow us concluding whether the microbiome of the C57BL/6J and BALB/c can be tailored by the host genotypes. In addition, recording of the cytokines levels defining the Th1/Th2 immune answer in serum and of the calprotectin concentration in the cecal content will correlate possible phenotypic outcomes with the gut microbiome. Overall, these aspects are essential for different pathologies where microbiota seem to be involved and a contribution of host genetic is assumed. |
TAKEN: Selection of suitable reference genomes for downstream analysis of bacterial cohorts |
|
 |
The enormous success and ubiquitous application of next and third generation sequencing has led to a large number of available high-quality draft and complete microbial genomes in the public databases. Today, the NCBI RefSeq database contains ~16,000 complete bacterial genomes. Concurrently, the selection of appropriate reference genomes (RGs) is increasingly important as it has enormous implications for routine in-silico analyses, as for example in detection of single nucleotide polymorphisms, scaffolding of draft assemblies, comparative genomics, etc. To address this issue many databases, methods and tools have been published in recent years e.g. RefSeq, DNA-DNA hybridization, average nucleotide identity (ANI) as well as percentage of conserved DNA values and kmer hashing methods (Mash). Nevertheless, the sheer amount of currently available databases and potential RGs contained therein, together with the plethora of tools available, often requires manual selection of the most suitable RGs. To tackle this issue the bioinformatics command line tool ReferenceSeeker (https://github.com/oschwengers/referenceseeker) was recently designed and implemented which combines a fast kmer profile-based lookup of candidate reference genomes (CRGs) from high quality databases with rapid computation of (mutual) highly specific ANI and conserved DNA values in a scalable and rapid implementation. As the analysis of cohorts of microbial genomes becomes increasingly important (viral & bacterial outbreaks), this approach should be extended from now providing the m best RGs for a single query genome to the best m RGs for a cohort of n query genomes. Therefore, new scoring metrics and ranking methods need to be tested and evaluated. In addition, a precise assessment of the impact of the RG selection on subsequent analyses (SNP detection & creation of phylogenetic trees) is an interesting question which deserves further attention. |
TAKEN: QC 16S trimming |
|
 |
16S NGS data often have not been trimmed, or were uploaded to ENA untrimmed. Thus, when imported to Qiita, the user shall be warned about this situation. How to detect it? Map fragments to rep set of GG and check if leading mismatches occur. |
TAKEN: Grammar to SVG |
|
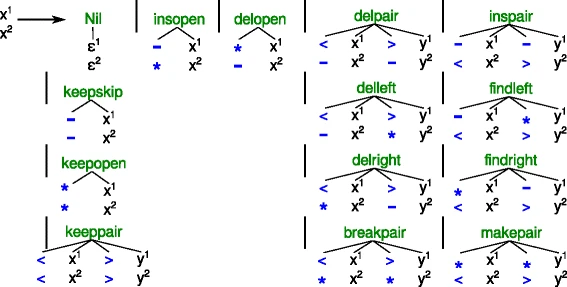 |
We think of the search space of an optimization Problem in ADP in terms of a tree grammar. These can be drawn as collections of little trees / forests and is a great way to communicate design decisions to others. However, for the actual computer program, we need to translate these drawing into something the machine can handle, i.e. ASCII text. To keep program and design in sync, it would be greate extend our compiler gapc such that it can directly generate SVG graphics from the ASCII version of the tree grammars. |
TAKEN: CLI for RNAhybrid 3.0 |
|
 |
The program RNAhybrid predicts potential targets for miRNAs. It was written in the early days of ADP in Haskell (version 1) and was ported into C via ADPc (version 2) some 18 years ago. It is time to lift this program into Bellman's GAP with new energy parameters, temperature modifications, fixes to the underlying grammar, and many more algorithmic tricks. Besides the core algorithm, we need a modern command line interface (CLI) - written in Python, acompanied with a rich test bed and high quality documentation. |

Additional open thesis topics are offered by our partner lab of Prof. Dr. Alexander Goesmann for Bioinformatics & Systems Biology. You will find open topics here.